

新修订《药品管理法》将于2019年12月1日起施行,相关配套的《药品注册管理办法(修订草案征求意见稿)》、《药品生产监督管理办法(修订草案征求意见稿)》、《药品经营监督管理办法(征求意见稿)》也已在近日发布。
药品注册是影响药品上市的一大法规方向,本文主要分析注册分类和注册审评时限缩短方面对行业的影响。
1
注册分类如何变化?
2015年版看点:
化学药五类注册框架
自2015年“722”以来,化学药品注册分类发生改变,分为5类,分别为:国内创新药、国内改良型新药、国内仿制药、进口药(其中包括两类:境外已上市境内未上市化学药、进口仿制药)。
2017年版看点:
生物制品分类变化
2017年10月23日,原CFDA先后发布《药品注册管理办法(修订稿)》和《〈中华人民共和国药品管理法〉修正案(草案征求意见稿)》的征求意见稿,主要针对临床研究审批“宽进严出”,明确药品的责任主体是药品上市许可持有人,对药品管理采取全链条和全生命周期管理的。但是,由于药品上市许可持有人制度的试点延期,所以当时征求意见稿所提到的中药和生物制品的改革分类,一直没有正式稿。
2017年《药品注册管理办法(修订稿)》公布的生物制品和中药、天然药物分类,基本与此前公布的化学药品分类方法相似,都是以创新药、改良药和仿制药为主要的基础框架体系进行分类。
中药、天然药物单独设定了个新3类古代经典名方。
生物制品方面,2017年《药品注册管理办法(修订稿)》分为预防用生物制品和治疗用生物制品。嵌合抗原受体T细胞(CAR-T)、自然杀伤细胞(naturalkiller cell,NK)和靶向活化自然杀伤细胞(taNKCell)等国际前沿的免疫细胞疗法属于“治疗用生物制品”。全新的基因治疗和细胞治疗类生物制品(例如创新机理、新载体、新靶细胞等)属于注册分类1类。在境内外已上市制品基础上进行改进的基因治疗和细胞治疗类生物制品,则属于注册分类2类。
生物制品的进口药分类与当时化学药品的进口药分类略有不同。当时的化学药品将进口药根据原研/仿制分为“5.1境外上市的原研药品(包括原料药及其制剂)申请在境内上市”和“5.2境外上市的非原研药品(包括原料药及其制剂)申请在境内上市”。2017年《药品注册管理办法(修订稿)》中,生物制品则还是沿用创新药、改良药和仿制药分类方法,预防用生物制品的新5类“进口疫苗”分为“5.1新型疫苗”、“5.2改良型疫苗”、“5.3境外上市、境内未上市的疫苗”和“5.4境内已上市的疫苗”四类。治疗用生物制品的新5类则分为“5.1新型生物制品”、“5.2改良型生物制品”、“5.3境外上市、境内未上市的生物制品”和“5.4境内已上市的生物制品”。
2019年版看点:
进口、国产仿制药合一
刚刚发布的《药品注册管理办法(修订草案征求意见稿)》,其基础架构为创新药、改良型新药、仿制药和境外已上市境内未上市药。
其中,中药的仿制药预计为“同方类似药”,并且继续增设“古代经典名方中药复方制剂”。
化学药注册分类将进口仿制药和国产仿制药合二为一,这意味着进口仿制药将和国产仿制药的审评标准统一,审批时排队的序列也会面临统一。进口仿制药以往所选择的参比制剂往往是选择当地上市的原研药,这可能会面临补充研究。
生物制品并没有仿制药的说法,只有生物类似药。《药品注册管理办法(修订草案征求意见稿)》则增设“境内已上市生物制品(包括生物类似药和不按生物类似药管理的境内已上市生物制品)”。境内未上市的生物制品的生物类似药,预计属于“境外已上市境内未上市生物制品”,但仍待相关细则的公布。
2
非处方药机会来了!
《药品注册管理办法(修订草案征求意见稿)》还优化了非处方药注册。
四种情况可直接申请注册
以下四种情形可直接提出非处方药上市注册:(一)国内已有相同活性成分、适应症或者功能主治、剂型、规格的非处方药上市的药品;(二)经国家局确定的非处方药改变剂型或者规格,但不改变适应症或者功能主治、给药剂量以及给药途径的药品;(三)使用国家局确定的非处方药的活性成分组成的新的复方制剂;(四)其他直接申报非处方药的情形。符合上述情形之一的,可直接提出非处方药上市注册。但从修订草案征求意见稿难以得知“其他直接申报非处方药的情形”有哪些情形。
儿童药OTC引力大
OTC分类的放开,预计会推动企业研发“使用国家局确定的非处方药的活性成分组成的新的复方制剂”以及“经国家局确定的非处方药改变剂型或者规格,但不改变适应症或者功能主治、给药剂量以及给药途径的药品”。特别是后者,“符合儿童生理特征的儿童用药品新品种、剂型和规格”可以优先审评缩短药品注册时间,将成为企业开发儿科药的主要方向。
国家局药品审评中心将非处方药的申报转国家局药品评价中心进行非处方药适宜性审查。非处方药适宜性审核时限为30个工作日,相对于药物临床试验申请、药物临床试验期间补充申请的审评时限60个工作日(含审批时限)以及药物临床试验所需要的时间,非处方药的注册审核时限较短,也是吸引企业愿意布局的一个原因。
3
药械组合“擦边球”被封
值得注意的是,拟申报注册的药械组合产品,已有同类产品经属性界定为药品的,按药品进行申报;尚未经属性界定的,申请人应当在申报注册前向国家局申请产品属性界定。属性界定为药品为主的,按照本办法规定的程序进行注册,其中属于器械部分的研究资料由国家器审中心作出审评结论后,转交国家局药品审评中心进行综合审评。例如过往含药用成分以敷料申报一类器械快速上市但实际上是透皮贴剂的,未来可能都会归属于药品,“擦边球”的门路将会被封死。
4
注册快道谁更快?
能否豁免或者通过优先审评减免一定的临床研究上市,是行业企业所关注的。临床豁免利好的主要是化学药仿制药,优先审评主要利好创新药。
针对化学仿制药的豁免
化学仿制药经申请人评估,认为无需或者不能开展药物临床试验的,申请人可提出豁免药物临床试验直接申请药品上市注册。但是,豁免药物临床试验的指导原则,由国家局药品审评中心另行制定发布,该指导原则也尚未公布。
针对创新药的四个程序比较

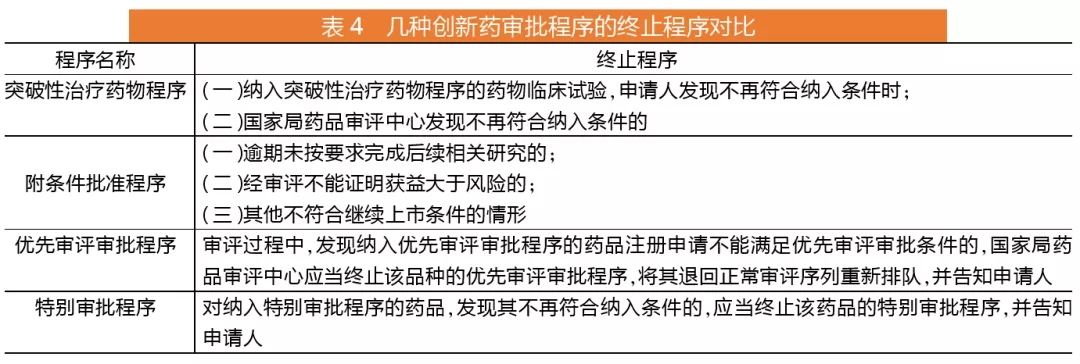
创新药可以给予技术指导、全过程沟通、优先配置资源、缩短审评时限等政策支持。从目前在实施的“附条件批准”和“优先审评”政策变成四个程序,分别为突破性治疗药物程序、附条件批准程序、优先审评审批程序及特别审批程序。

其中,突破性治疗药物程序主要针对药品临床试验阶段,优先审评审批则明确了对应药品上市注册申请阶段。实际上,真正能缩短审评时长的是优先审评审批程序和特别审批程序,优先审评审批程序的药品上市注册审评时限120个工作日;临床急需的境外已上市的罕见病药品审评时限60个工作日;特别审批程序的审评时限暂未明确。

★★★ 小结 ★★★
《药品注册管理办法(修订草案征求意见稿)》将进口仿制药和国产仿制药放在同一条起跑线,预计仿制药的竞争强度会进一步加大。
非处方药法规的放开,将推动个别企业开发非处方药。但是,非处方药所处的领域都是竞争激烈的治疗领域,企业开发非处方药要更多地考虑渠道覆盖和品牌营销能力。在药店市场,同适应症的独家产品拼的是品牌力。
对于新药来说,优先审评是上市注册缩短的最关键程序。利好的领域没有特别的变化,创新疫苗特别是肿瘤疫苗也许会是一大突破点。
专利链接和新药监测期在此前3月修订的药品管理办法实施条例已有体现,专利纠纷需要参考上位法,但专利法暂无修订的意向,预计将维持现状。
(来源于:医药经济报 原创: 边界)